
European Journal of Neurodegenerative Diseases 2024; 13(1) January-April: 1-7
INVESTIGATION OF ADRENERGIC MODULATION ON SHOCK INDUCED BY PLATELET ACTIVATING FACTOR
C. Iaccarino 1* and R. Galzio 2
1 Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy;
2 Neurosurgery Unit, Department of Surgical Sciences, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy.
*Correspondence to:
Dr. Corrado Iaccarino,
Department of Biomedical, Metabolic and Neural Sciences,
University of Modena and Reggio Emilia,
41125 Modena, Italy.
e-mail: Iaccarino.corrado@gmail.com
| Received: 26 February, 2024 Accepted: 18 April, 2024 |
2974-6345 (2024) Copyright © by BIOLIFE This publication and/or article is for individual use only and may not be further reproduced without written permission from the copyright holder. Unauthorized reproduction may result in financial and other penalties. Disclosure: all authors report no conflicts of interest relevant to this article. |
ABSTRACT
Platelet activating factor (PAF) is a lipid mediator which is released by the immune system during inflammatory processes and causes an anaphylactoid reaction when it is administered in rodents. Two models which have been widely used to investigate the systemic pathophysiology of PAF are PAF-induced hypotension in rats and PAF-induced death in mice. Utilizing these models, studies have suggested that the sympathetic nervous system plays a key role in modulating the detrimental effects of PAF. In this study, we sought to systematically evaluate the effects of adrenergic blockade and pharmacological augmentation of adrenergic reflexes on PAF-induced hypotension and death in rodents. Intravenous PAF administration in rats caused profound systemic arterial hypotension affecting the heart which was associated with bradycardia, suggesting an inability of the rats to compensate for the decreased arterial pressure. The beta blocker propranolol also significantly potentiated the mortality of intravenous administration of PAF in mice. In contrast to the beta-adrenergic system, alpha-adrenergic mechanisms appear less integral in the adaptive response to the PAF challenge. In rats, phentolamine, an alpha-adrenergic blocker, prolonged the hypotension provoked by PAF, but heart rate remained responsive. This effect suggest that cardiac reflexes were intact, and phentolamine had no effect on PAF-induced lethality in mice. Naloxone and thyrotropin releasing hormone (TRH) are two drugs which augment sympathetic responses, and these were also tested against PAF-induced death in mice. Both drugs were protective against PAF lethality, providing further evidence of the adaptive role of the sympathetic system against systemic PAF. Therefore, these studies support the concept that beta-adrenergic mechansims are an essential component for compensatory responses to systemic anaphylactoid responses involving PAF, and suggest a potentially lethal effect of beta blockers during systemic immune or inflammatory reactions involving PAF. This demonstrates that the beta-adrenergic system is a necessary component for the adaptive response to systemic PAF.
KEYWORDS: PAF, anaphylactic shock, thyrotropin releasing hormone, propranolol, naloxone, phentolamine
INTRODUCTION
Platelet activating factor (PAF) (1-0-alkyl 2-acetyl sn glyceryl phosphoryl choline) is released by leukocytes, platelets, vascular endothelium, and other tissue and is a potent mediator of immune and inflammatory reactions (1). Its major systemic effects include promotion of vascular permeability, bronchoconstriction, hypotension, and above all, platelet aggregation. In vivo studies have demonstrated a role for PAF in diverse physiological and pathological states, including IgE-induced systemic anaphylaxis (2), endotoxin shock (3,4), IgG-induced shock (5), and transplant rejection (6).
Different models exist for studying the systemic action of PAF, and one which is simple and useful is PAF-induced mortality in mice (7-10). In this model, the intravenous injection of PAF causes dose-dependent hypotension, hemoconcentration, and death. The lethality of i.v. PAF in mice is thought to be due to the combination of the effects shock and bronchoconstriction, but not thrombosis. This is because platelets of rodents are not directly responsive to PAF (11), and so, PAF toxicity in mice has been proposed as a model for lethal anaphylactic shock. The mortality in this model can be prevented by pretreatment with steroidal anti-inflammatory drugs such as cortisone, inotropic drugs, PAF antagonists, or specific antibodies against PAF, and is exacerbated by beta-adrenergic blocking drugs (specifically beta-2 antagonists) (12-14).
The effects of adrenergic manipulation in this model are very interesting, since the sympathetic nervous system plays a central role in the response to circulatory collapse in shock. Since PAF is an important mediator of anaphylactic and septic shock, it activates the sympathetic nervous system, and adrenergic blockade should prove deleterious in PAF-induced shock. In fact, the i.v. infusion of PAF in rats results in elevated plasma catecholamine levels (15), and beta blockers, in addition to enhancing the lethality of PAF in mice, also slow the recovery of blood pressure to normal levels after the PAF challenge. In contrast to the harmful effects of beta blockade, thyrotropin releasing hormone (TRH), which stimulates sympathetic outflow and catecholamine release, has been shown to reverse PAF-induced hypotension in guinea pigs (16).
These observations implicate the sympathetic nervous system as a mechanism for modulating the systemic effects of PAF in rodents. The PAF- adrenergic interaction should be further studied, focusing on the role of alpha-adrenergic mechanisms, to determine whether, and to what extent, enhanced sympathetic outflow can improve survival and hemodynamics in rodents challenged with PAF. The main goals of this study were to systematically evaluate the effects of alpha- and beta-adrenergic blockers, and to compare the effects of these blockers with drugs which stimulate or enhance sympathetic outflow in PAF-induced death in mice. In addition to TRH, naloxone was also tested, as it enhances sympathetic outflow and thus also improves hemodynamic recovery in some shock models.
MATERIALS AND METHODS
PAF-induced hypotension in rats
Male Wistar rats, weighing approximately 225 g, were purchased from Charles River Laboratories and were randomly assigned to control and experimental groups. Rats were anesthetized with sodium pentobarbital (60 mg/kg i.p.) and placed on a heating pad with a rectal temperature probe to maintain body temperature above 37.5°C. The right carotid artery and jugular vein were cannulated for recording arterial blood pressure and for injecting drugs, respectively. Propranolol, phentolamine (2.5 mg/kg each), or the 0.9% NaCl vehicle was injected, followed by the injection of PAF (2 μg/kg) or its vehicle (0.9% NaCl) after three minutes. The dose of PAF was selected as one which produces profound but reversible hypotension. Both the adrenergic blockers and PAF were dissolved at concentrations which resulted in injection volumes of 1 ml/kg body weight. Each of the four PAF-challenged groups (propranolol or phentolamine pretreatment groups and the two respective vehicle-pretreated control groups) included 10 rats, whereas the drug-only control groups consisted of 7 animals.
Blood pressure was monitored continuously for the following 30 min by a P23 ID pressure transducer and transducer amplifier and recorder. Heart rate (HR) was measured each minute. For each adrenergic blocker (propranolol or phentolamine) and its two associated control groups (vehicle pretreated, PAF challenged group and drug pretreated, vehicle challenged group), mean arterial pressure (MAP) and HR were compared between the three groups by one-way analysis of variance (ANOVA). When the ANOVA indicated statistically significant between-group differences, it was followed by specific comparison of the adrenergic blocker pretreated, PAF-challenged group to each of the two control groups by the Bonferroni method. P<0.05 was considered to be statistically significant.
PAF toxicity in mice
Male CD-1 mice, weighing approximately 25 g, were obtained from Charles River Laboratories and assigned randomly to the groups below. Mice were anesthetized with sodium amytal (100 mg/kg ip). Propranolol (2.5 mg/kg, Sigma Chemical Co.), phentolamine (2.5 mg/kg, Ciba Pharmaceutical Co.) or the combination of both drugs (2.5 mg/kg each) was then administered into the jugular vein. Drugs were dissolved in 0.9% NaCl at a concentration of 0.5 mg/ml, resulting in an injection volume of 5 μl/g body weight. For each of these treatment groups, a control group of mice received an injection of the 0.9% NaCl vehicle. Initially, each experimental and control group consisted of 15 mice, but the propranolol group and its control were expanded to 40 mice to confirm initial positive findings. Five minutes after drug or vehicle pretreatment, an approximate LD50 of PAF (15 μg/kg, iv) was administered. Mice were then observed until either death or full recovery of the righting reflex, which occurred within 4 hours in all cases. PAF (Calbiochem™) was prepared in 0.9% NaCl solution at a concentration resulting in an injection volume of 5 μl/g body weight to achieve the desired dose. Mortality was compared between drug and vehicle treated groups by the Chi square test.
Additional groups of 15 mice were pretreated with TRH (2 mg/kg, i.v.), naloxone (2 mg/kg, i.v.), or the NaCl vehicle 2 min before the PAF challenge. TRH and naloxone were tested against the approximate PAF LD80 (40 μg/kg) and TRH was also tested using the lower dose of PAF (15 μg/kg). Again, all drugs and the vehicle were dissolved in 0.9% NaCl and were administered at injection volumes of 5 μl/g body weight. The Chi square test was used to compare the mortality between the drug pretreatment and control groups.
RESULTS
PAF-induced hypotension in rats
The injection of 2.0 μg/kg i.v. PAF resulted in severe, rapidly developing hypotension, with only partial recovery of MAP within 30 min. However, HR was unaffected by PAF in vehicle-pretreated rats. The combination of propranolol and PAF produced a peak hypotensive response similar to PAF alone (vehicle/PAF group), but MAP was significantly depressed in the propranolol/PAF rats relative to the vehicle/PAF control group during the recovery period at several time points. HR was markedly different in these two groups, with significantly lower HR in the propranolol-pretreatment group for the duration of the study. Notably, propranolol alone produced a similar effect on HR and caused mild hypotension as well.
Like propranolol, phentolamine treatment alone significantly reduced blood pressure, and phentolamine in combination with PAF resulted in lower MAP during the recovery period compared to vehicle-pretreated rats. The alpha- adrenergic blocking drug by itself also resulted in a prolonged reduction in HR. Immediately following the PAF challenge, MAP was significantly lower in the phentolamine/PAF group compared to the vehicle/PAF rats (P<0.05); this comparison was also significant at later time points. Although HR was diminished by phentolamine for the duration of the study in the vehicle-challenged group, it was significantly reduced in the phentolamine/PAF rats (relative to the vehicle/PAF group) only immediately prior to PAF injection and in the first 2 min after the challenge. HR then recovered rapidly to control levels. The two phentolamine-treated groups also had different MAP levels before the challenge with PAF or vehicle (P<0.05).
PAF toxicity in mice
Propranolol pretreatment significantly exacerbated the acute toxicity of i.v. PAF in mice. Injection of the approximate LD50 of PAF (based on previous studies) in the propranolol-treated group resulted in a significantly greater mortality rate than the same challenge in the control group (P<0.01) (Fig. 1). On the other hand, phentolamine pretreatment had no significant effect on PAF toxicity (Fig. 1). The combination of propranolol and phentolamine produced effects similar to propranolol alone, with a higher mortality rate than the control group (P<0.01) (Fig. 1).
TRH exerted a protective effect against both the approximate PAF LD80 (P<0.01) and LD50 (P<0.05) (Fig. 2). Similarly, pretreatment with naloxone significantly reduced the lethality of the high dose PAF challenge (P<0.05) (Fig. 2).
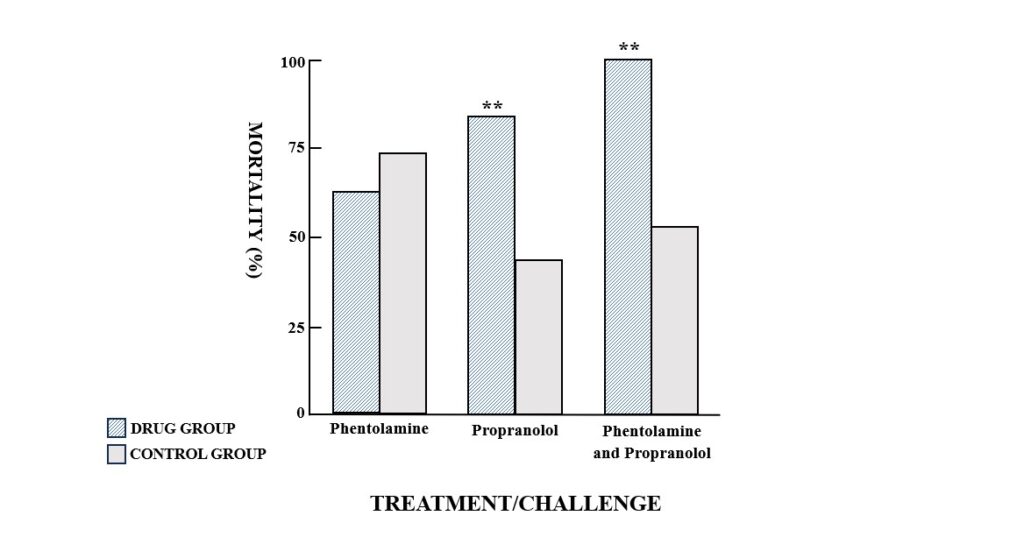
Fig. 1. Mortality in groups of mice challenged with an approximate LD50 of platelet activating factor (PAF) (15μg/kg, i.v.). Mice were pretreated i.v. with either the alpha-adrenergic antagonist phentolamine (2.5 mg/kg), the beta-adrenergic antagonist propranolol (2.5mg/kg) or a combination of both drugs (2.5 mg/kg each) prior to PAF administration. Control mice received a 0.9% NaCl injection before the PAF challenge. **P<0.01, compared to the appropriate control group.
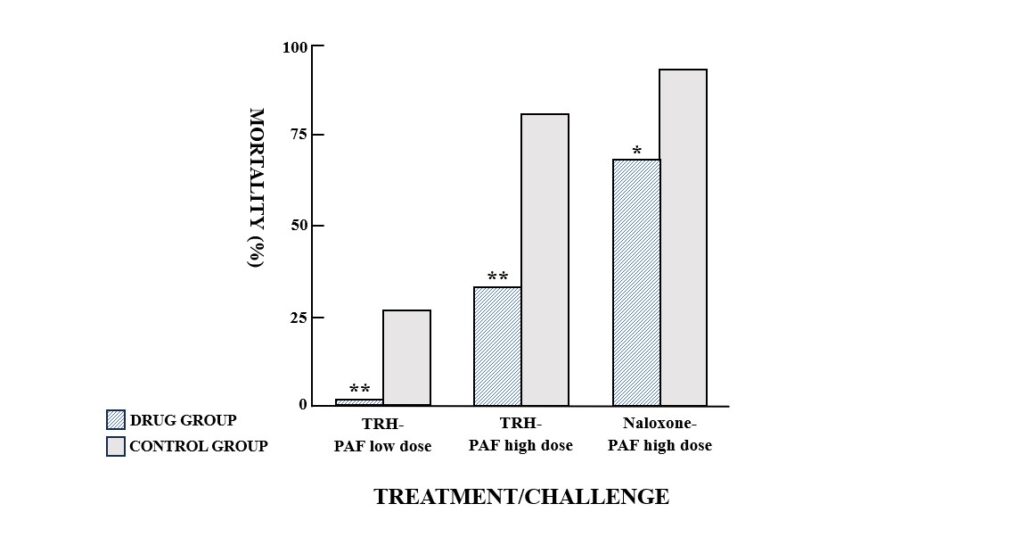
Fig. 2. Mortality in groups of mice challenged with platelet activating factor (PAF) (low dose, 15 μg/kg, i.v., or high dose, 40 μg/kg, i.v.). Mice were pretreated i.v. with thyrotropin releasing hormone (TRH) (2 mg/kg, i.v.) or naloxone (2 mg/kg, i.v.) prior to PAF administration. Control mice received a 0.9% NaCl injection before the PAF challenge. *P<0.05, **P<0.01, compared to the appropriate control group.
DISCUSSION
A harmful interaction has been suggested between adrenergic blocking drugs and PAF in shock. PAF administration elevates plasma levels of catecholamines in the rat, probably due to a sympathetic reflex provoked by hypotension (15). Intravenous or intracerebroventricular injection of TRH activates the sympathetic nervous system through a central nervous system (CNS) mechanism and reverses PAF-induced hypotension in guinea pigs (16). Beta-adrenergic blockers potentiate PAF toxicity in mouse models, an effect which is probably mediated by beta-2 receptors based on differential effects of selective beta blockers (17). It has been speculated that the bronchopulmonary effects of the beta blockers increased PAF mortality.
The results of the experiments presented here shed light on the interaction between adrenergic mechanisms and PAF in shock states and support the important role that the sympathetic nervous system plays in counteracting the effects of PAF in anaphylactic reactions. These studies clearly demonstrate that in one model (PAF toxicity in mice), the two drugs which increase sympathetic outflow through different mechanisms (TRH and naloxone) and the beta-adrenergic blocker propranolol have opposite effects on PAF lethality. Enhancement of sympathetic reflexes is apparently protective against lethal shock, whereas blockade of the beta-adrenergic component of the sympathetic system is harmful.
The present studies also suggest the lack of significant involvement of alpha-adrenergic processes in modulating acute responses to PAF. Unlike propranolol, phentolamine did not increase the lethality of PAF in mice. Because of this result, and previous studies which showed that the adrenergic system and PAF included beta blockers and adrenal demedullation but not alpha blockers, the effects of both phentolamine and propranolol on PAF-induced alterations in MAP and HR in rats were examined in the current experiments. These hemodynamic studies also confirm the importance of beta, rather than alpha, adrenergic mechanisms in modulating the acute effects of PAF and suggest that the cardiovascular and bronchopulmonary system are involved in the potentiation of PAF toxicity by beta blockers.
In line with previous studies, propranolol in combination with PAF provoked a more prolonged hypotension than PAF alone in the anesthetized preparation used here. Propranolol itself produced persistent bradycardia and a moderate fall in MAP; this effect might contribute to the delay in recovery from PAF-induced hypotension in the propranolol/PAF group. The combination of PAF and propranolol resulted in an initial fall in MAP to the same level as PAF without propranolol, but the prolonged hypotensive phase was accompanied by bradycardia when propranolol was present. These results differ from those observed in the conscious rat, in which PAF promoted tachycardia which was blocked by propranolol. In the anesthetized rat, there is most likely significant adrenergic tone, prior to the administration of any drug. It is likely that bradycardia in the presence of hypotension reflects an inability of the propranolol-treated rats to respond to the PAF-induced fall in MAP, which is related largely to peripheral vasodilation by increasing cardiac output (18). On this basis, the actions of propranolol in the mouse model could be due to cardiovascular effects of the beta blocker, in addition to the bronchopulmonary action (17). the protective actions of positive inotropic drugs against PAF support the idea that cardiovascular dysfunction is involved in the lethal actions of PAF in mice.
Phentolamine, like propranolol, produced bradycardia and hypotension independent of PAF administration. No particular significance is attached to the marginally significant difference in MAP in the two phentolamine-treated groups before PAF or vehicle challenge. The difference might have been related to the slightly different basal MAP levels in the two groups. Phentolamine-pretreated rats had depressed MAP immediately after the challenge with PAF and during the recovery phase, compared to saline-pretreated PAF-challenged animals. The prolonged hypotension probably reflects the vasodilatory actions of both phentolamine and PAF, and the inhibition of compensatory (alpha-adrenergic) vasoconstriction. The bradycardia associated with phentolamine treatment was quickly reversed following the PAF challenge, but was persistent in the phentolamine-treated, saline-challenged group. This suggests that although PAF-induced hypotension is prolonged under alpha-adrenergic blockade, the cardiac compensatory, beta-adrenergic mechanisms are maintained and result in increased HR and, possibly, enhanced cardiac output.
CONCLUSIONS
This study lends support to the idea that beta-adrenergic mechanisms are an adaptive and crucial component of the response to the anaphylactoid effects of PAF. However, alpha-adrenergic mechanisms seem to be less significant in the acute response to PAF-induced effects. Finally, because beta-adrenergic function is important in the recovery from PAF, the combination of propranolol and PAF results in a potentially lethal interaction, while agents which increase adrenergic systems protect against PAF. PAF-induced mortality and hypotension in rodents could help to explain the sympathetic responses in pathological states of immunity.
Statement of ethics
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding sources
Not applicable.
REFERENCES
- Weyrich AS, Lindemann S, Zimmerman GA. The evolving role of platelets in inflammation. Journal of Thrombosis and Haemostasis. 2003;1(9):1897-1905. doi:https://doi.org/10.1046/j.1538-7836.2003.00304.x
- Finkelman FD, Rothenberg ME, Brandt EB, Morris SC, Strait RT. Molecular mechanisms of anaphylaxis: Lessons from studies with murine models. Journal of Allergy and Clinical Immunology. 2005;115(3):449-457. doi:https://doi.org/10.1016/j.jaci.2004.12.1125
- Jeong YI, Jung ID, Lee CM, et al. The Novel Role of Platelet-Activating Factor in Protecting Mice against Lipopolysaccharide-Induced Endotoxic Shock. Pockley G, ed. PLoS ONE. 2009;4(8):e6503. doi:https://doi.org/10.1371/journal.pone.0006503
- Sun XM, Hsueh W. Platelet-activating factor produces shock, in vivo complement activation, and tissue injury in mice. The Journal of immunology. 1991;147(2):509-514. doi:https://doi.org/10.4049/jimmunol.147.2.509
- Strait RT, Morris SC, Finkelman FD. IgG-blocking antibodies inhibit IgE-mediated anaphylaxis in vivo through both antigen interception and Fc RIIb cross-linking. Journal of Clinical Investigation. 2006;116(3):833-841. doi:https://doi.org/10.1172/jci25575
- Makowka L, Chapman FA, Cramer DV, Qian SG, Sun H, Starzl TE. Platelet-activating factor and hyperacute rejection. Transplantation. 1990;50(3):359-365. doi:https://doi.org/10.1097/00007890-199009000-00001
- Lu J, Pierce M, Franklin A, Jilling T, Stafforini DM, Caplan M. Dual Roles of Endogenous Platelet-Activating Factor Acetylhydrolase in a Murine Model of Necrotizing Enterocolitis. Pediatric Research. 2010;68(3):225-230. doi:https://doi.org/10.1203/pdr.0b013e3181eb2efe
- Abhilasha KV, Sumanth MS, Chaithra VH, et al. p38 MAP-kinase inhibitor protects against platelet-activating factor-induced death in mice. Free radical biology & medicine. 2019;143:275-287. doi:https://doi.org/10.1016/j.freeradbiomed.2019.08.019
- Amorim CZ, Cordeiro RS, Vargaftig BB. Involvement of platelet-activating factor in death following anaphylactic shock in boosted and in unboosted mice. European journal of pharmacology. 1993;235(1):17-22. doi:https://doi.org/10.1016/0014-2999(93)90814-x
- Fukuda Y, Kawashima H, Saito K, Inomata N, Matsui M, Nakanishi T. Effect of human plasma-type platelet-activating factor acetylhydrolase in two anaphylactic shock models. European journal of pharmacology. 2000;390(1-2):203-207. doi:https://doi.org/10.1016/s0014-2999(99)00920-6
- Klee A, Schmid-Schönbein GW, Seiffge D. Effects of platelet activating factor on rat platelets in vivo. European journal of pharmacology. 1991;209(3):223-230. doi:https://doi.org/10.1016/0014-2999(91)90173-n
- Upton J, Vadas P. Potential Therapeutic Strategies for Severe Anaphylaxis Targeting Platelet-Activating Factor and PAF Acetylhydrolase. Current Treatment Options in Allergy. 2014;1(3):232-246. doi:https://doi.org/10.1007/s40521-014-0020-2
- Ishii S, Kuwaki T, Nagase T, et al. Impaired Anaphylactic Responses with Intact Sensitivity to Endotoxin in Mice Lacking a Platelet-activating Factor Receptor. Journal of Experimental Medicine. 1998;187(11):1779-1788. doi:https://doi.org/10.1084/jem.187.11.1779
- Koltai M, Hosford D, Guinot P, Esanu A, Braquet P. Platelet Activating Factor (PAF). Drugs. 1991;42(1):9-29. doi:https://doi.org/10.2165/00003495-199142010-00002
- Brimijoin S, Hammond P, Khraibi AA, Tyce GM. Catecholamine Release and Excretion in Rats with Immunologically Induced Preganglionic Sympathectomy. Journal of Neurochemistry. 2008;62(6):2195-2204. doi:https://doi.org/10.1046/j.1471-4159.1994.62062195.x
- Feuerstein G, Lux WE Jr, Snyder F, Ezra D, Faden AI. Hypotension produced by platelet-activating factor is reversed by thyrotropin-releasing hormone. Circulatory shock. 1984;13(3):255-260.
- Criscuoli M, Subissi A. Paf‐acether‐induced death in mice: involvement of arachidonate metabolites and β‐adrenoceptors. British journal of pharmacology. 1987;90(1):203-209. doi:https://doi.org/10.1111/j.1476-5381.1987.tb16841.x
- Lai FM, Shepherd CA, Cervoni P, Wissner A. Hypotensive and vasodilatory activity of (±) 1-0-octadecyl-2-acetyl glyceryl-3-phosphorylcholine in the normotensive rat. Life sciences. 1983;32(10):1159-1166. doi:https://doi.org/10.1016/0024-3205(83)90122-4